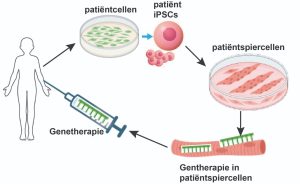
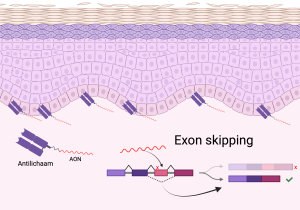
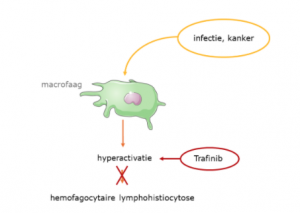
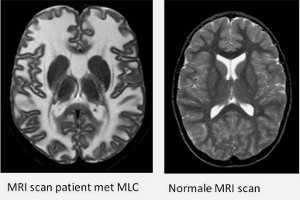
Lopende onderzoeken
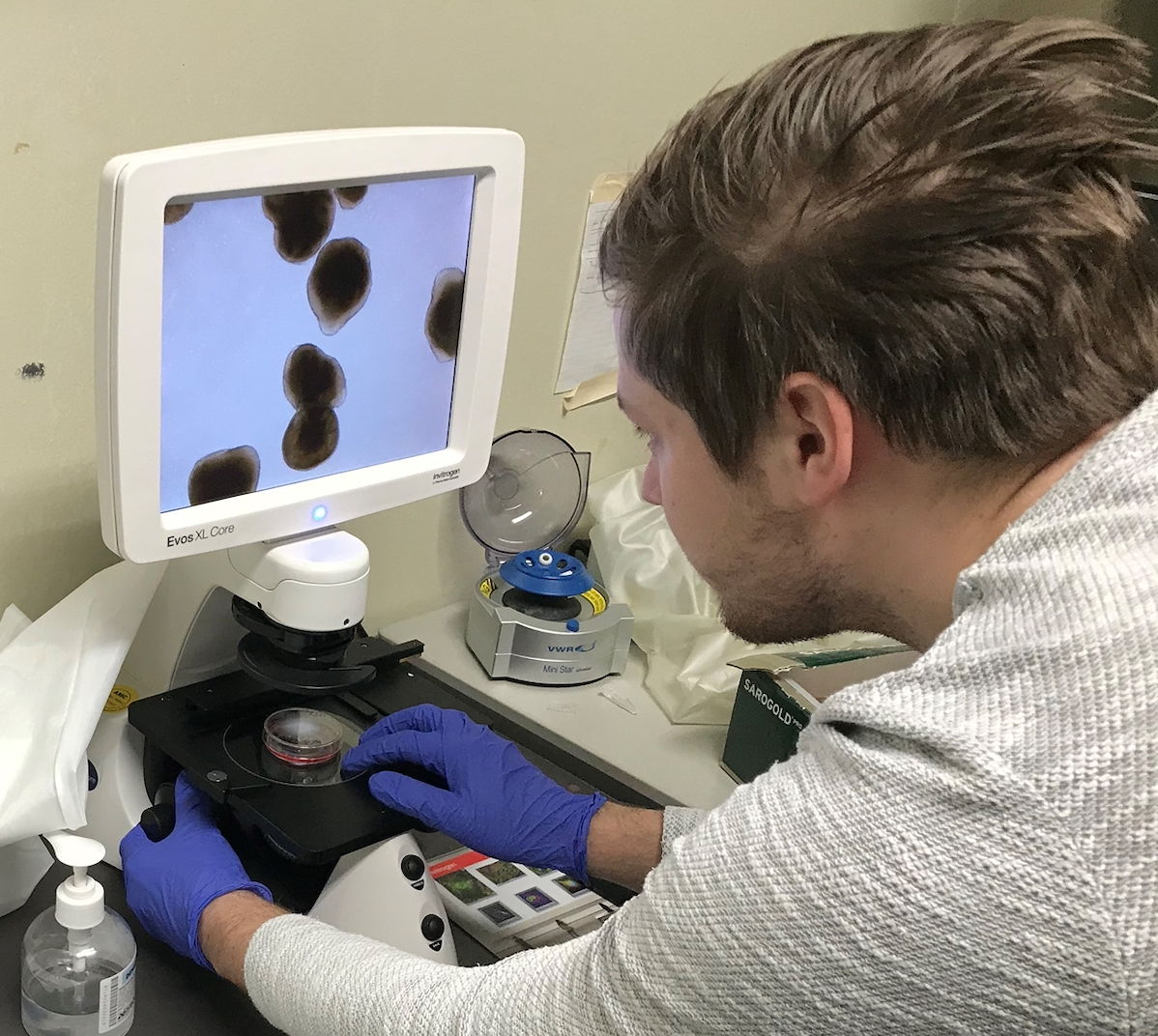
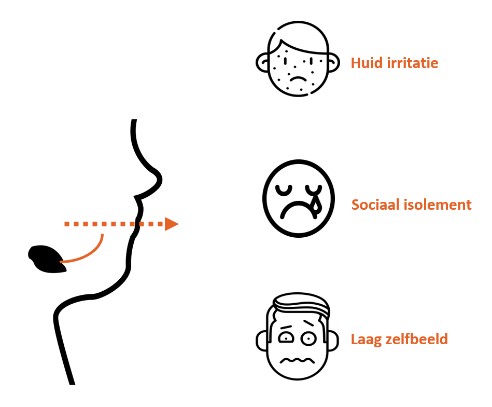
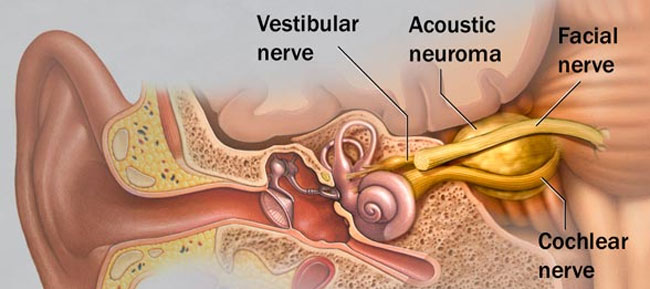
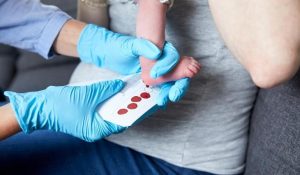
Hieronder vindt u een aantal van de lopende onderzoeken die momenteel plaats vinden met door het ZZF en sponsoren gefinancierde bijdrages. Niet alle lopende onderzoeken kunnen we hier plaatsen. Heeft u een specifieke vraag, schroom niet om ons te bellen of mailen.